Wprowadzenie
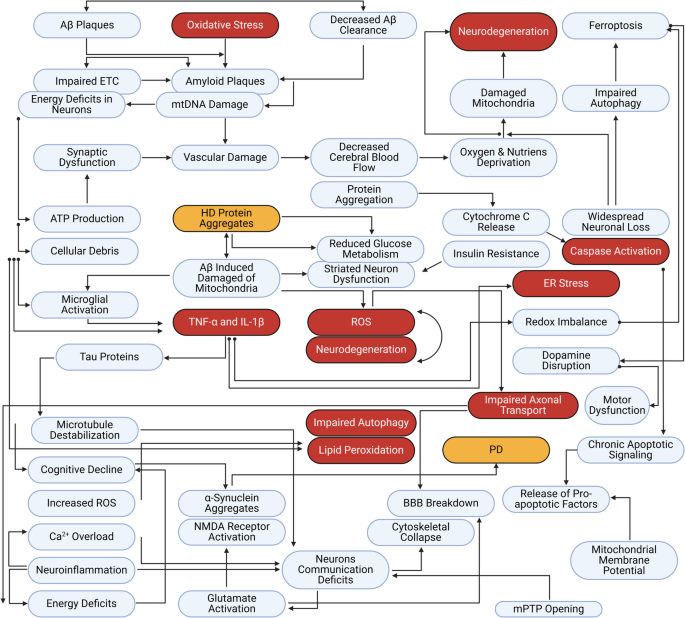
Neurodegeneracja w chorobach takich jak choroba Alzheimera (AD), choroba Parkinsona (PD) czy stwardnienie zanikowe boczne (SLA) wiąże się z postępującą utratą neuronów i funkcji mózgu. Wobec ograniczeń terapii objawowych, rośnie zainteresowanie podejściami neuroprotekcyjnymi – czyli takimi, które chronią neurony przed uszkodzeniem lub opóźniają ich degenerację. Peptydy neuroprotekcyjne to krótkie łańcuchy aminokwasów wykazujące zdolność do wspierania przeżycia komórek nerwowych i hamowania procesów neurodegeneracyjnych. Wiele endogennych neuropeptydów (np. grelina, neuropeptyd Y, PACAP, substancja P, neurotensyna) ma udokumentowane działanie ochronne w modelach chorób neurodegeneracyjnych – m.in. zapobiegają aktywacji kaspazy-3 (apoptozie neuronów), zmniejszają stres oksydacyjny mitochondriów, hamują aktywację mikrogleju oraz regulują autofagię. Oprócz naturalnych neuropeptydów, opracowywane są także syntetyczne peptydy terapeutyczne, które mogą celować w konkretne szlaki patogenetyczne (np. hamować agregację toksycznych białek). Niniejszy artykuł przedstawia definicję i klasyfikację peptydów neuroprotekcyjnych, molekularne mechanizmy ich działania, przykłady kluczowych peptydów tej klasy oraz aktualne kierunki badań przedklinicznych i klinicznych, a także potencjalne zastosowania i wyzwania związane z terapią peptydową.
Definicja i klasyfikacja peptydów neuroprotekcyjnych
Peptydy neuroprotekcyjne definiuje się jako peptydy (typowo o długości kilkunastu do kilkudziesięciu aminokwasów), które potrafią zapobiegać uszkodzeniu neuronów lub wspierać ich przeżycie w obliczu czynników patologicznych. Można je podzielić na kilka kategorii:
- Endogenne neuropeptydy o działaniu troficznym – np. peptydy z rodziny neurotrofin (połowa neurotrofin, takich jak BDNF, to duże białka, ale istnieją też krótsze analogi peptydowe), peptydy hormonalne (np. IGF-1, somatomedyna C), czy neuropeptydy regulujące metabolizm (np. GLP-1 – peptyd glukagonopodobny 1). Należą tu także neuropeptydy układu opioidowego (endorfiny, enkefaliny, dynorfiny) pełniące funkcje neuromodulatorów.
- Neuropeptydy sensu stricto – krótkie peptydy przekaźnikowe uwalniane przez neurony lub komórki glejowe, oddziałujące na receptory metabotropowe (GPCR) i wykazujące działanie ochronne. Przykłady: PACAP (pituitary adenylate cyclase-activating polypeptide) i pokrewna VIP (vasoactive intestinal peptide), neuropeptyd Y (NPY), grelina, substancja P, neurotensyna i inne, które w OUN mogą modulować przeżycie neuronów.
- Peptydy mitochondrialne – niewielkie peptydy kodowane przez genom mitochondrialny, odkryte stosunkowo niedawno. Najlepiej poznanym jest humanin (24-aminokwasowy peptyd) oraz spokrewnione peptydy podobne do humanin (SHLP1-6) i MOTS-c. Wykazują one szerokie działanie cytoprotekcyjne, w tym neuroprotekcyjne.
- Fragmenty peptydowe białek neuroprotekcyjnych – niewielkie domeny peptydowe wyizolowane z większych białek o działaniu troficznym lub ochronnym. Np. NAP (davunetide) – 8-aminokwasowy fragment białka ADNP; krótki peptyd semafosporowy P5 (TFP5) pochodzący z aktywatora kinazy CDK5; fragmenty pochodne BDNF czy GDNF, które zachowują aktywność biologiczną.
- Syntetyczne peptydy terapeutyczne – zaprojektowane de novo lub udoskonalone analogi naturalnych peptydów. Mogą działać jako agoniści receptorów (np. mimetyki BDNF aktywujące receptor TrkB) lub jako inhibitory szkodliwych procesów (np. peptydy zapobiegające agregacji amyloidu-beta, alfa-synukleiny czy białka Tau). Przykładowo opracowano liczne peptydy antyagregacyjne dla AD: sekwencje beta-breaker wnikające w struktury amyloidu (np. iAβ5p – Ac-Leu-Pro-Phe-Phe-Asp-NH_2), peptydy α-helikalne i α-sheet wiążące formy oligomeryczne Aβ, cykliczne peptydy blokujące oddziaływania patologiczne białek itp..
W kolejnych rozdziałach omówiono mechanizmy działania i przykłady najważniejszych peptydów z powyższych kategorii w kontekście AD, PD, SLA i pokrewnych chorób.
Mechanizmy działania na poziomie molekularnym i komórkowym
Peptydy neuroprotekcyjne wykazują zróżnicowane mechanizmy działania, często angażując szlaki sygnałowe odpowiedzialne za przeżycie komórek i homeostazę. Kluczowe mechanizmy obejmują:
- Aktywacja receptorów promujących przeżycie neuronów: Wiele peptydów działa poprzez receptory błonowe – np. neurotrofiny i ich analogi aktywują receptory tyrozynowe Trk, co uruchamia kaskady PI3K/Akt/mTOR oraz MAPK/ERK prowadzące do ekspresji genów pro-przeżyciowych (jak BCL-2, CREB) i hamowania sygnałów apoptotycznych. Przykładowo, peptydowy agonista receptora TrkB naśladujący BDNF indukuje fosforylację Akt i ERK w neuronach, co sprzyja ich przetrwaniu. Z kolei PACAP i VIP oddziałują na receptory GPCR (PAC1, VPAC1/2), aktywując cAMP/PKA oraz szlaki PKC, MAPK i PI3K/Akt, które konwergują na hamowanie kaskady apoptotycznej (np. blokadę aktywacji kaspazy-3).
- Działanie antyapoptotyczne: Wiele peptydów zwiększa ekspresję białek antyapoptotycznych lub hamuje proapoptotyczne. Dla przykładu, DADLE – analog endogennej enkefaliny działający na receptory δ-opioidowe – w modelu komórkowym PD znacząco poprawiał przeżywalność neuronów dopaminergicznych poddanych stresowi retikulum endoplazmatycznego. Mechanistycznie DADLE obniżał poziom sensorów odpowiedzi na nieprawidłowe białka (UPR) i agregatów białkowych, pobudzał fosforylację kinazy MEK/ERK, zwiększał ekspresję czynnika wzrostu nerwów NGF oraz białka Bcl-2, a hamował ekspresję proapoptotycznego BIM. Efektem netto była silna ochrona komórek przed śmiercią apoptotyczną. Podobnie PACAP poprzez receptory PAC1/VPAC potrafi zwiększać poziom Bcl-2 i zmniejszać aktywację kaspaz, chroniąc neurony ruchowe przed apoptozą w modelach SLA.
- Hamowanie stresu oksydacyjnego i dysfunkcji mitochondrialnej: Liczne peptydy wykazują właściwości antyoksydacyjne lub wspierają funkcje mitochondriów. Przykładowo, humanin oraz jego analogi chronią komórki przed toksycznością amyloidu-beta m.in. przez neutralizację wolnych rodników i poprawę funkcji łańcucha oddechowego. Humanin może bezpośrednio wiązać się z proapoptotycznym czynnikiem Bax w mitochondrium, zapobiegając jego oligomeryzacji i uwolnieniu cytochromu c, co blokuje inicjację apoptozy. Inny syntetyczny peptyd SS-31 (elamipretid) jest celowany do mitochondriów i stabilizuje kardiolipinę w wewnętrznej błonie mitochondrialnej, redukując powstawanie ROS – wykazano, że zmniejsza uszkodzenia oksydacyjne neuronów w modelach AD i chorób siatkówki.
- Przeciwzapalne modulowanie mikrogleju: Niektóre neuropeptydy tłumią przewlekły stan zapalny w OUN. Np. neuropeptyd Y działając poprzez receptory Y1/Y2 hamuje aktywację prozapalnych mikrogleju i astrogleju, obniżając wydzielanie cytokin prozapalnych, co pośrednio chroni neurony przed uszkodzeniem. Podobnie VIP i PACAP mają udokumentowane właściwości immunomodulujące – zmniejszają uwalnianie TNF-α, IL-6 oraz ograniczają naciek komórek zapalnych w mózgu.
- Hamowanie agregacji patologicznych białek: Unikalnym mechanizmem syntetycznych peptydów jest ich zdolność do specyficznego wiązania i neutralizacji toksycznych form białek odkładających się w mózgu. Przykładowo, krótki peptyd LPFFD (sekwencja zawarta w iAβ5p) może wbudowywać się w strukturę amyloidu-β i zaburzać jego konformację, hamując dalsze tworzenie oligomerów i włókien. Analogicznie, opracowano peptydy kierowane przeciw α-synukleinie (w PD) – np. peptyd AFF wiążący centralny region NAC α-synukleiny, co zapobiega jej agregacji i toksyczności w modelach komórkowych. Innym podejściem jest stosowanie peptydów katabolicznych – sekwencji aktywujących degradację danych białek (np. peptydy promujące autofagię lub proteasomalne usuwanie zmutowanych białek, jak zaobserwowano dla analogu PACAP w SBMA).
Warto podkreślić, że mechanizmy te często nakładają się na siebie – jeden peptyd może wykazywać działanie plejotropowe. Przykładowo PACAP jednocześnie aktywuje kaskady pro-przeżyciowe (PKA/AKT), działa antyapoptotycznie i przeciwzapalnie, a ponadto w pewnych modelach sprzyja neurogenezie i regeneracji aksonów. Takie wielotorowe działanie czyni peptydy obiecującymi kandydatami na leki modyfikujące przebieg choroby (DMT) w neurologii.
Przykłady wybranych peptydów neuroprotekcyjnych
Peptydy mitochondrialne: humanin i analogi
Jednym z pierwszych odkrytych peptydów mitochondrialnych o działaniu neuroprotekcyjnym był humanin – 24-aminokwasowy peptyd zidentyfikowany w 2001 r. w bibliotekach cDNA neuronów u pacjentów z AD. Okazał się on potężnym czynnikiem chroniącym neurony przed toksycznością amyloidu-beta oraz przed uszkodzeniami wywołanymi mutacjami rodzinnymi AD (APP, PSEN1). Późniejsze badania wykazały, że humanin jest kodowany przez mitochondrialne DNA (w ramach 16S rRNA) – co zapoczątkowało koncepcję mitochondrialnie kodowanych peptydów (MDP) jako nowych endogennych regulatorów homeostazy komórkowej.
Humanin działa zarówno wewnątrzkomórkowo, jak i poprzez receptory błonowe. W cytoplazmie i mitochondrium potrafi bezpośrednio oddziaływać z proapoptotycznymi białkami Bax i Bak, zapobiegając ich oligomeryzacji i wywołaniu apoptozy mitochondrialnej. Ponadto wykazano, że humanin wiąże się z IGFBP-3 (białkiem wiążącym IGF-1) oraz z receptorami powiązanymi z gp130 na powierzchni komórek, aktywując szlaki przetrwania zależne od STAT3 i ERK. W efekcie humanin zwiększa odporność neuronów na różne stresory – od β-amyloidu po stres oksydacyjny i ischemiczny. Poziomy humanin endogennie spadają z wiekiem i w chorobach neurodegeneracyjnych (np. AD), co sugeruje, że niedobór tego peptydu może predysponować do uszkodzeń neuronów. Trwają prace nad analogami humanin o zwiększonej stabilności i sile działania – np. HNG (humanin z zamianą S14G) jest kilkadziesiąt razy silniejszy niż natywny peptyd w ochronie neuronów przed amyloidem. Inne mitochondrialne peptydy, takie jak MOTS-c (16 aa, wpływa na metabolizm glukozy) czy SHLP2 (peptyd podobny do humanin), również wykazują potencjał neuroprotekcyjny poprzez wspomaganie funkcji mitochondrialnych i redukcję stanu zapalnego. Ogólnie, peptydy MDP stanowią nowy wymiar endogennej obrony przed neurodegeneracją i są intensywnie badane jako możliwe terapie spowalniające starzenie układu nerwowego.
Pochodne BDNF i peptydy neurotroficzne
Brain-Derived Neurotrophic Factor (BDNF) jest kluczowym białkiem troficznym wspierającym neurony, ale jego bezpośrednie zastosowanie terapeutyczne jest utrudnione (duża cząsteczka ~27 kDa, słabo przenika do OUN). Dlatego opracowuje się mimetyki peptydowe BDNF – mniejsze cząsteczki zdolne aktywować receptor TrkB i naśladować pro-neurotropowe działanie BDNF. Strategią jest często wykorzystanie sekwencji z pętli BDNF odpowiadających za wiązanie z TrkB, połączenie dwóch takich sekwencji dla zwiększenia awidności lub chemiczna modyfikacja w celu poprawy stabilności. Przykładem są opracowane w Rosji dimeryczne dipeptydy na bazie pętli 4 BDNF: GSB-106 (bis-[monosuccinylo-L-seryl-L-lysyna] heksametylenodiamid) oraz nowszy GSB-214. GSB-106 działa jako partialny agonista TrkB – w badaniach in vivo podawany przewlekle wykazał działanie przeciwdepresyjne (odwracał objawy depresji u myszy poddanych stresowi przewlekłemu) oraz wspomagał regenerację po udarze (zapobiegał deficytom poznawczym i depresyjnym u szczurów po niedokrwieniu mózgu). Z kolei GSB-214 okazał się czynnym agonistą TrkB o działaniu neuroprotekcyjnym w modelu PD – w rotencyjnym modelu 6-OHDA podanie GSB-214 poprawiło koordynację ruchową i zmniejszyło apoptozę neuronów w prążkowiu, co powiązano z aktywacją kinaz Akt i MAPK w tych neuronach. Co ważne, peptyd ten przekracza barierę krew-mózg po podaniu obwodowym, choć jego biodostępność doustna pozostaje niska ze względu na trawienie enzymatyczne. Innym podejściem są peptydomimetyki o strukturze nietypowej – np. cyklopeptyd TDP6 oparty na domenie pętli 2 BDNF, czy też małe cząsteczki niepeptydowe (np. 7,8-DHF) działające na TrkB. Większość z nich wykazała skuteczność w przedklinicznych modelach AD/PD, choć próby kliniczne napotkały trudności (część związków nie przeszła faz z powodu nieskuteczności lub działań niepożądanych). Niemniej nowa generacja mimetyków (np. związki firmy AlzeCure – ACD855, ACD856 – modulatory allosteryczne Trk) weszła do badań klinicznych we wczesnym AD.
Warto wspomnieć, że podobne wysiłki dotyczą innych neurotrofin: NGF (czynnik wzrostu nerwów) – tutaj przykładem jest dipeptyd GK-2 aktywujący TrkA, czy Ciproreksyna (peptyd z pętli 1 NGF). Dla NT-3 i NT-4 również powstały analogi peptydowe. Celem tych badań jest ominięcie ograniczeń pełnowymiarowych neurotrofin (krótki T<sub>1/2</sub>, efekty uboczne jak allodynia) i stworzenie bezpiecznych, selektywnych neurotrophin mimetics zdolnych do spowolnienia neurodegeneracji. Pierwsze tego typu leki są na etapie testów – np. LM11A-31 (mała cząsteczka aktywująca p75^NTR/Trk, faza II w AD) czy wspomniany ACD856 (pozytywny modulator Trk w fazie I).
Peptydy układu opioidowego o działaniu neuroprotekcyjnym
Endogenne peptydy opioidowe – encefaliny, endorfiny, dynorfiny – znane są głównie z modulacji przewodzenia bólu i emocji, lecz odgrywają również rolę w regulacji przeżycia neuronów. Szczególnie interesujący jest szlak receptora δ-opioidowego (DOR), którego aktywacja okazała się sprzyjać przeżyciu neuronów w warunkach stresu. D-Ala^2, D-Leu^5 enkefalina (DADLE) – stabilny analog met-enkefaliny – wykazał intrygujące działanie ochronne w różnych modelach. Wspomniano wyżej jego wpływ na zahamowanie UPR i apoptozy w komórkach PD. Ponadto, w modelach zwierzęcych DADLE oraz inne agonisty DOR zmniejszały uszkodzenia neurologiczne w udarze mózgu i w urazach niedokrwienno-reperfuzyjnych poprzez aktywację szlaku PKC/MAPK oraz indukcję autofagii ochronnej. W modelu MPTP (toksyczne uszkodzenie neuronów dopaminergicznych) podanie agonistów δ-opioidowych ograniczało utratę neuronów istoty czarnej, co wiązano m.in. z redukcją stresu ER, obniżeniem stężenia Ca^2+ w cytoplazmie i zwiększeniem czynników troficznych w mózgu. W przeciwieństwie do receptorów μ i κ, których długotrwała stymulacja może być szkodliwa, sygnalizacja δ-wydaje się “przełączać” neurony w tryb wyciszenia ekscytotoksycznego i stresowego. Wykorzystanie peptydów opioidowych w terapii neurodegeneracji jest jednak wyzwaniem – dawki muszą być tak dobrane, by uniknąć działań niepożądanych (sedacji, tolerancji). Trwają prace nad selektywnymi agonistami δ-opioidowymi pozbawionymi działania uzależniającego. Niemniej układ opioidowy jawi się jako interesujący cel: np. niedawno wykazano, że pochodne enkefaliny mogą zmniejszać dyskinezy wywołane L-DOPĄ w PD poprzez modulację plastyczności synaptycznej w prążkowiu, co sugeruje wielowymiarowe korzyści potencjalnej terapii peptydami opioidowymi.
PACAP – plejotropowy neuropeptyd o działaniu ochronnym
PACAP (Pituitary Adenylate Cyclase-Activating Polypeptide) to 38-aminokwasowy neuropeptyd (istnieje też krótsza forma 27-aa) zaangażowany w liczne funkcje rozwojowe i homeostatyczne układu nerwowego. Receptory PACAP (PAC1 specyficzny dla PACAP oraz VPAC1/2 wspólne z VIP) są szeroko eksprymowane w OUN. Od odkrycia PACAP w 1989 r. jako hormonu przysadkowego, wykazano jego silne działanie neurotroficzne i neuroprotekcyjne w wielu modelach chorób neurodegeneracyjnych.
W AD stwierdzono, że PACAP chroni neurony przed toksycznością amyloidu-beta – np. podanie PACAP myszom transgenicznym APP/PS1 spowalniało tworzenie blaszek i poprawiało pamięć przestrzenną. Mechanizmy obejmują hamowanie prozapalnej aktywacji mikrogleju, obniżenie fosforylacji Tau oraz bezpośrednie efekty antyapoptotyczne w neuronach hipokampa. W modelach PD PACAP również wykazuje działanie ochronne na neurony dopaminergiczne – potrafi np. zwiększać przeżycie neuronów w hodowlach traktowanych toksynami parkinsonowskimi poprzez aktywację szlaku PKA/CREB i czynnika NRF2 (redukcja stresu oksydacyjnego). Co ważne, poziom endogennego PACAP może maleć w stanach chorobowych – w mózgach pacjentów z PD i ALS obserwowano zmniejszoną ekspresję PACAP i jego receptora, co może przyczyniać się do podatności neuronów ruchowych na degenerację.
W SLA i pokrewnych chorobach neuronu ruchowego (MND) PACAP okazał się szczególnie obiecujący. U myszy z mutacją SOD1 (model ALS) delecja genu PACAP przyspieszała utratę neuronów autonomicznych, natomiast leczenie PACAP-em in vitro zwiększało przeżywalność motoneuronów z IPS po głodzeniu, poprzez aktywację kinazy PKA, receptorów EGFR i szlaku MMP-2. Co więcej, PACAP wpływa na patomechanizmy specyficzne – np. w modelu SBMA (choroba Kennedy’ego, z patologiczną ekspansją poliglutaminy w receptorze androgenowym) PACAP zmniejsza fosforylację i akumulację zmutowanego receptora, ułatwiając jego degradację, co przynosiło ochronę neuronów ruchowych. W badaniu Polanco i wsp. (2016) podanie nowego, stabilnego analogu PACAP myszom SBMA od początku objawów wydłużyło ich przeżycie i poprawiło funkcje ruchowe, zmniejszając poziom zlogregowanych białek w rdzeniu. Rezultaty te potwierdzają potencjał PACAP jako kandydat na terapię modyfikującą chorobę. Aktualnie trwają prace nad analogami PACAP o dłuższym czasie półtrwania i większej selektywności (np. zmodyfikowane formy nie wywołujące efektów naczyniorozszerzających charakterystycznych dla VIP).
Oprócz bezpośredniej neuroprotekcji, PACAP może działać neuroregeneracyjnie. W modelach uszkodzeń OUN (np. uraz rdzenia) egzogenny PACAP stymuluje odrost aksonów i nasila działanie komórek macierzystych. Wykazuje także synergizm z innymi terapiami – np. w połączeniu z komórkami macierzystymi mezenchymalnymi potęguje ich efekt naprawczy w uszkodzeniu rdzenia. Te wielokierunkowe właściwości czynią PACAP jednym z najintensywniej badanych peptydów – określa się go wręcz jako “endogenny strażnik neuronów”.
NAP (davunetide) – peptyd chroniący mikrotubule
NAP (znany też jako davunetide lub AL-108) to 8-aminokwasowy fragment (NAPVSIPQ) pochodzący z Activity-Dependent Neuroprotective Protein (ADNP) – białka o właściwościach neurotroficznych, odgrywającego rolę w rozwoju mózgu i stabilizacji mikrotubul. Odkrywczyni ADNP, Illana Gozes, wykazała że NAP odtwarza wiele funkcji ochronnych pełnowymiarowego białka. Mechanizm działania NAP polega głównie na stabilizacji cytoszkieletu neuronów: wiąże się on z końcami mikrotubul (przez interakcję z białkami EB1/EB3) i zapobiega ich depolimeryzacji pod wpływem czynników uszkadzających. Ponieważ agregacja hiperfosforylowanego białka Tau prowadzi do dysfunkcji mikrotubul w AD i innych tauopatiach, NAP może przeciwdziałać temu procesowi. Rzeczywiście, w hodowlach neuronalnych NAP chronił mikrotubule przed toksycznością peptydu β-amyloidu, a w modelach mysiich z nadmierną fosforylacją Tau zmniejszał odkładanie patologicznego białka i poprawiał funkcje poznawcze. Co więcej, w mysim modelu ciężkiej neurodegeneracji NAP poprawiał transport aksonalny i hamował zwyrodnienie aksonów.
W modelach in vivo NAP wykazuje szeroki profil neuroprotekcyjny: u myszy z częściową delecją ADNP (Adnp^+/-), które mają deficyty pamięci, osłabione mięśnie i przyspieszone obumieranie neuronów, leczenie peptydem NAP (CP201) znacząco poprawiło ich zdolności poznawcze, funkcje ruchowe oraz zmniejszyło neurodegenerację. To wskazuje, że NAP potrafi kompensować utratę funkcji ADNP in vivo.
Ze względu na obiecujące wyniki przedkliniczne, davunetide został przetestowany klinicznie jako potencjalna terapia chorób neurodegeneracyjnych, zwłaszcza tauopatii. W niewielkim badaniu u osób z łagodnymi zaburzeniami poznawczymi zauważono poprawę uwagi i pamięci roboczej po 12 tygodniach stosowania NAP donosowo. Największą próbą była jednak faza 2/3 w postępującym porażeniu nadjądrowym (PSP) – ciężkiej tauopatii. Niestety, pomimo dobrego bezpieczeństwa, roczne leczenie davunetidem (30 mg donosowo 2x dziennie) nie spowolniło objawów PSP w porównaniu z placebo. Wyniki opublikowane w Lancet Neurology (2014) wykazały brak różnic w zmianie skal oceny PSP i codziennych czynności między grupami NAP a placebo. Interpretowano to jako dowód, że sama stabilizacja mikrotubul może nie wystarczyć w zaawansowanej tauopatii lub że konieczne są silniejsze analogi peptydu. Mimo tego niepowodzenia klinicznego, NAP pozostaje aktywnie badany – zwłaszcza w kontekście zespołu ADNP, rzadkiego zaburzenia ze spektrum autyzmu spowodowanego haploinsuficjencją ADNP. W modelach komórkowych mutacji ADNP wykazano, że NAP przenika nawet do jądra komórkowego i natychmiast koryguje nieprawidłową lokalizację zmutowanego białka ADNP, przywracając prawidłową morfologię jądra. To unikalne działanie sugeruje, że NAP może mieć właściwości regulujące ekspresję genów i stabilność genomu. Ze względu na dobre bezpieczeństwo (próby kliniczne nie wykazały istotnych skutków ubocznych poza podrażnieniem nosa), planowane są dalsze badania NAP jako terapii dzieci z mutacjami ADNP.
Inne syntetyczne peptydy w badaniach
W literaturze opisano wiele dodatkowych peptydów o działaniu neuroprotekcyjnym, które znajdują się na etapie badań przedklinicznych:
- Peptydy przeciw amyloidowi i Tau: Poza wspomnianymi iAβ5p i analogami NAP, testuje się np. peptydy CI-PTD i D3 wiążące oligomery Aβ, peptydy Tau-5 zapobiegające agregacji białka Tau, a także peptydy destabilizujące α-synukleinę. Przykładowo, racjonalnie zaprojektowany 9-aa peptyd PDpep ograniczył tworzenie się fibrilli α-synukleiny i chronił komórki przed jej cytotoksycznością. Tego typu inhibitory agregacji mogą działać bardzo specyficznie, ale wyzwaniem jest ich dostarczenie do neuronów w mózgu.
- Peptydy antyoksydacyjne i antyapoptotyczne: Wspomniany SS-31 przeszedł już fazy I/II w niewydolności mitochondrialnej, a jego działanie w modelach AD budzi nadzieje na spowalnianie zmian neurodegeneracyjnych poprzez ochronę mitochondriów. Inny krótki peptyd HNG17 (pochodna humanin) testowany jest pod kątem udaru mózgu – w modelach zwierzęcych zmniejszał on wielkość zawału i deficyty neurologiczne po udarze.
- Peptydy przenikające do mózgu (BBB-shuttle peptides): Aby zwiększyć skuteczność terapii peptydowych, trwają prace nad nośnikami i modyfikacjami zwiększającymi przenikalność bariery krew-mózg. Do niektórych kandydujących peptydów dodaje się sekwencje przenikające (np. TAT z HIV) lub wykorzystuje nanocząstki peptydowe. Przykładem kandydata leczniczego jest nerinetyd (znany też jako Tat-NR2B9c lub NA-1) – peptyd hamujący interakcję białka PSD-95 z receptorami NMDA, zapobiegając ekscytotoksyczności. W dużym badaniu klinicznym u pacjentów z ostrym udarem niedokrwiennym (ESCAPE-NA1, 2020) nerinetyd poprawił rokowanie u chorych nieleczonych trombolizą, choć efekt zanikał przy jednoczesnym podaniu tPA (enzym ten degradował peptyd). Mimo to, nerinetyd stanowi dowód, że peptydy mogą być bezpiecznie stosowane u ludzi w stanach ostrej neuroprotekcji. Obecnie prowadzi się dalsze analizy, jak zoptymalizować jego działanie.
Badania przedkliniczne i kliniczne – aktualny stan i perspektywy
W ostatnich latach obserwuje się intensyfikację badań nad peptydami w terapiach chorób neurodegeneracyjnych. W badaniach przedklinicznych pojawiają się kolejne dowody skuteczności peptydów w modelach zwierzęcych AD, PD, SLA i innych. Przykładowo, analogi GLP-1 (liraglutyd, eksenatyd) poprawiały parametry pamięci u myszy z amyloidozą AD, obniżając poziom Aβ i fosfo-Tau w mózgu. Peptydowy agonista receptorów ghreliny – relina – zmniejszał utratę neuronów dopaminowych i objawy ruchowe u modeli PD poprzez aktywację szlaku AMPK i biogenezy mitochondriów.
W badaniach klinicznych kilka peptydów osiągnęło fazy oceniające skuteczność:
- Analogi inkretyn (GLP-1): Wykorzystanie leków cukrzycowych okazało się obiecujące w neurodegeneracji. Eksenatyd (agonista GLP-1) w pilotażowym badaniu fazy II w chorobie Parkinsona wykazał sygnał potencjalnego spowolnienia pogorszenia – po 12 miesiącach pacjenci leczeni eksenatydem mieli nieznacznie lepsze wyniki motoryczne (MDS-UPDRS) niż grupa placebo, a poprawa utrzymywała się nawet po odstawieniu leku. Chociaż duża próba fazy III (EXENATIDE-PD3, ~200 pacjentów) nie potwierdziła istotnego statystycznie spowolnienia progresji PD, analiza podgrup sugeruje, że wcześniejsze stadia choroby mogą bardziej korzystać z terapii. Obecnie inne analogi GLP-1 są testowane: liraglutyd w chorobie Alzheimera (badanie ELAD – ocena wpływu na akumulację amyloidu i funkcje poznawcze) oraz semaglutyd (faza III w wczesnym AD). Wstępne wyniki wskazują, że liraglutyd może poprawiać metabolizm mózgowy i niektóre funkcje poznawcze u chorych na AD. Trwa także badanie LIXIPARK oceniające lixisenatyd u pacjentów z wczesną chorobą Parkinsona – w fazie II odnotowano wolniejszy przyrost niepełnosprawności ruchowej w ciągu 12 mies. w grupie lixisenatydu vs placebo. Agoniści GLP-1 są o tyle atrakcyjni, że mają dopuszczenie do użytku (w cukrzycy) i znany profil bezpieczeństwa – droga do ich ewentualnej repurposing w neurologii jest więc krótsza.
- IGF-1 (insulinopodobny czynnik wzrostu): Ze względu na silne działanie neurotroficzne IGF-1 próbowano w SLA. Dwa duże badania kliniczne (lata 90.) z podskórnym rhIGF-1 nie wykazały jednak istotnych korzyści klinicznych, a metaanaliza w 2012 r. wskazała co najwyżej minimalne wydłużenie przeżycia bez poprawy siły mięśni. Przyczyną porażki mogło być słabe przenikanie IGF-1 do OUN i obwodowe efekty uboczne. Podjęto więc próby alternatywne: w 2005 r. mała próba podania IGF-1 dokanałowo 9 chorym na SLA dała obiecujące rezultaty (poprawa funkcji ruchowych), a u zwierząt PEGylacja IGF-1 poprawiła dystrybucję i efekty terapeutyczne. Obecnie badane są wektory AAV dostarczające IGF-1 do OUN. Mimo trudności, IGF-1 pozostaje ważnym „starym znajomym” w kontekście neuronów ruchowych, a jego modyfikowane formy (np. NOPALS – fragment ťažący cząstkę sygnałową IGF-1) mogą pojawić się w przyszłości.
- Inne peptydy w badaniach klinicznych: W kontekście udaru wspomniano nerinetyd – niestety jego zastosowanie okazało się zależne od braku trombolizy. Cerebrolizyna – mieszanina peptydów z mózgu świń – jest stosowana w niektórych krajach w demencji i udarze; meta-analizy sugerują umiarkowaną poprawę funkcji poznawczych u pacjentów z łagodną do umiarkowanej AD, choć dowody nie są jednoznaczne i preparat ten budzi kontrowersje co do mechanizmu (zawiera m.in. fragmenty analogiczne do BDNF, GDNF). W chorobie Huntingtona trwają próby z peptydami inhibitorowymi hamującymi interakcje zmutowanej huntingtyny – np. peptyd P42 (fragment białka exon1 huntingtyny) poprawił fenotypy u myszy HD, a obecnie oceniany jest jego udoskonalony analog zdolny do przenikania do mózgu.
Ograniczenia i wyzwania terapii peptydowych
Mimo ekscytujących wyników badań, kliniczne wykorzystanie peptydów napotyka kilka istotnych wyzwań:
- Biodostępność i dostarczenie do mózgu: Peptydy podane obwodowo często ulegają szybkiemu rozkładowi przez peptydazy i mają krótki okres półtrwania. Ponadto większość z nich słabo przekracza barierę krew-mózg. W efekcie skuteczne dawki terapeutyczne mogą być wysokie. Rozwiązaniem są modyfikacje peptydów (np. podstawienie D-aminokwasów, cyklizacja, pegylacja) zwiększające oporność na enzymy i przenikalność. Wykorzystuje się także nośniki – nanocząstki, liposomy – oraz drogi podania omijające BBB (dooponowo, donosowo). Przykładowo, NAP podawany donosowo docierał do mózgu w stężeniu pozwalającym na efekt, choć wymagał aplikacji dwa razy dziennie. Skuteczne dostarczenie pozostaje jednak jednym z głównych ograniczeń.
- Stabilność i immunogenność: Peptydy mogą ulegać denaturacji lub agregacji podczas przechowywania, wymagają więc chłodzenia i ostrożnej formulacji. Dłuższe peptydy lub sekwencje pochodzenia nie-ludzkiego mogą wywoływać odpowiedź immunologiczną. Konieczne bywa usunięcie epitopów immunogennych lub stosowanie form lustrzanych (D-peptydów), które są obojętne immunologicznie. Z kolei D-peptydy mogą mieć problem z aktywacją celów biologicznych (receptory często preferują L-aminokwasy).
- Krótki czas działania: Z racji szybkiej eliminacji, wiele peptydów wymaga częstego podawania (np. codziennego wstrzykiwania). To rodzi problemy z compliance pacjentów, zwłaszcza w chorobach przewlekłych. Trwają prace nad formułami o przedłużonym uwalnianiu (implanety, pompy infuzyjne) lub prolekami. Przykładem rozwiązania jest stosowanie małych cząsteczek aktywujących te same szlaki co peptydy – np. 7,8-DHF jako „zamiennik” BDNF, czy małe mimetyki PACAP. Jednak na ogół brakuje im specyficzności peptydów i mogą mieć działania uboczne.
- Efekty uboczne systemowe: Peptydy o aktywności hormonalnej (jak IGF-1, GLP-1) mogą wywoływać działania niepożądane poza OUN. Np. agoniści GLP-1 powodują nudności, utratę masy ciała – co u chorych neurodegeneracyjnych (często z sarkopenią) wymaga ostrożności. IGF-1 niesie ryzyko hipoglikemii i potencjalnie stymulacji wzrostu nowotworów. Z tego względu poszukuje się sposobów celowania terapii do mózgu (np. przez wektory wirusowe selektywnie ekspresjonujące peptyd w OUN).
- Koszty i produkcja: Synteza peptydów leczniczych, zwłaszcza modyfikowanych czy cyklicznych, bywa kosztowna. Produkcja biologiczna (np. w bakteriach, drożdżach) dużych peptydów wymaga dodatkowych etapów oczyszczania i refoldingu. Niemniej, rozwój technik chemicznych i peptydowych drukarek obniża te koszty z roku na rok.
Mimo tych trudności, sukcesy niektórych peptydów (np. eksenatyd w pierwszych próbach PD, semaglutyd w AD, czy dawne wprowadzenie działającej neuroprotekcyjnie erytropoetyny modyfikowanej jako lek neuro, N-PEP) potwierdzają, że warto inwestować w ten kierunek. W obliczu starzenia się populacji i braku leków zatrzymujących neurodegenerację, terapie peptydowe stanowią obiecującą ścieżkę badawczą. Łączą one wysoką swoistość działania (jak leki biologiczne) z relatywnie niewielkim ryzykiem długotrwałych toksyczności (peptydy zwykle nie kumulują się, a rozkładają do aminokwasów). Przyszłość pokaże, czy ulepszone peptydy – być może dostarczane za pomocą nowoczesnych nośników – staną się pierwszymi skutecznymi lekami spowalniającymi choroby takie jak AD, PD czy SLA.
Bibliografia (publikacje 2018–2025)
- Zheng Y., Zhang L., Xie J., Shi L. The Emerging Role of Neuropeptides in Parkinson’s Disease. Front Aging Neurosci. 2021;13:646726.
- Zuccaro E., Piol D., Basso M., Pennuto M. Motor Neuron Diseases and Neuroprotective Peptides: A Closer Look to Neurons. Front Aging Neurosci. 2021;13:723871.
- Das S.K. et al. Protein and peptide based nanotherapeutics for the management of Alzheimer’s disease: Current insights and future directions. Ageing Res Rev. 2026;114:103000.
- Ibrahim R., Kambal A., Abdelmajeed M.A. Glucagon-Like Peptide-1 Receptor Agonists in Neurodegenerative Diseases: A Comprehensive Review. Cureus. 2025;17(9):e92441.
- Karachaliou C.E., Livaniou E. Neuroprotective Action of Humanin and Humanin Analogues: Research Findings and Perspectives. Biology (Basel). 2023;12(12):1534.
- Numakawa T., Kajihara R. The Role of BDNF as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection. Molecules. 2025;30(4):848.
- Moghal E.T.B., Venkatesh K., Sen D. The delta opioid peptide DADLE induces neuroprotection through cross-talk between the UPR and pro-survival MAPK-NGF-Bcl2 pathways via modulation of microRNAs under ER stress. Cell Biol Int. 2018;42(5):543-569.
- Gozes I. The ADNP Syndrome and CP201 (NAP) Potential and Hope. Front Neurol. 2020;11:608444.
- Boxer A.L. et al. Davunetide in patients with progressive supranuclear palsy: a randomized, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014;13(7):676-685.
- Solés-Tarrés I., Cabezas-Llobet N., Vaudry D., Xifró X. Protective Effects of PACAP and VIP Against Cognitive Decline in Neurodegenerative Diseases. Front Cell Neurosci. 2020;14:221.
Opracowanie: Rafał Klasiński / Fundacja Independent Institute of Chemical Processes