Streszczenie wykonawcze
Mitochondria odgrywają kluczową rolę w starzejącym się mózgu i patogenezie chorób neurodegeneracyjnych (Alzheimer, Parkinson, ALS, Huntington). Procesy mitochondriów – generacja ATP, buforowanie Ca²⁺ i regulacja apoptozy – ulegają zaburzeniu wraz z wiekiem, co prowadzi do spadku OXPHOS, wzrostu stresu oksydacyjnego i uszkodzeń DNA mitochondrialnego. W chorobach neurodegeneracyjnych opisano charakterystyczne deficyty łańcucha oddechowego (zwłaszcza kompleksów I i IV), nadprodukcję ROS oraz dysfunkcje dynamiki (fuzja/podział) i mitofagii. Dowody z badań komórkowych, zwierzęcych i pacjentów potwierdzają te mechanizmy: np. toksyny środowiskowe (MPTP, rotenon) hamują kompleks I i wywołują objawy choroby Parkinsona, a mutacje SOD1 czy TDP-43 w ALS prowadzą do utraty potencjału błonowego i uwalniania cytochromu c. Rozpoznaje się potencjalne markery mitochondrialne (np. poziomy SOD2, wolne mtDNA w płynie mózgowo-rdzeniowym). Interwencje terapeutyczne ukierunkowane na mitochondria – od celowanych antyoksydantów (MitoQ, SkQ1) i stymulatorów biogenezy (PGC-1α) po przeszczepy mitochondriów i terapie genowe – wykazują obiecujące efekty w modelach przedklinicznych. Kluczowe wyzwania pozostają jednak nierozwiązane: heterogeniczność defektów mitochondrialnych wśród pacjentów, trudności w dostarczeniu terapii do neuronów oraz brak przekrojowych badań klinicznych modyfikujących przebieg chorób (z wyjątkiem symptomatycznej terapii ALS). Niezbędne są dalsze, systematyczne badania w celu weryfikacji zależności przyczynowo-skutkowych i identyfikacji skutecznych biomarkerów mitochondriów (np. cf-mtDNA, SOD2).
Wstęp
Mózg jest narządem o bardzo wysokim zapotrzebowaniu na energię, a jego funkcjonowanie zależy w dużej mierze od metabolizmu mitochondrialnego. Mitochondria pełnią funkcję „elektrowni komórkowych” (produkcja ATP w OXPHOS), regulują homeostazę Ca²⁺, generują sygnały komórkowe i uczestniczą w mechanizmach śmierci komórki (apoptoza). Z wiekiem obserwuje się stopniowe pogorszenie funkcji mitochondriów: spada sprawność łańcucha oddechowego, zwiększa się poziom wolnych rodników (ROS), a mechanizmy naprawcze stają się niewystarczające. To pogorszenie homeostazy redoks i produkcji energii w istotny sposób przyczynia się do procesów starzenia się mózgu. Równocześnie, wiele chorób neurodegeneracyjnych wiąże się z charakterystycznymi defektami mitochondrialnymi: akumulacją uszkodzeń mtDNA, zaburzeniami dynamiki mitochondrialnej oraz dysfunkcją kontroli jakości organelli. Na przykład hipoteza „mitochondrialnego kaskadu alzheimerowskiego” sugeruje, że w AD deficyty mitochondrialne mogą zachodzić bardzo wcześnie i indukować dalsze patologie związane z β-amyloidem i tau (Di Lorenzo i in., 2025). Celem niniejszego raportu jest przegląd i analityczna synteza literatury naukowej dotyczącej ról mechanizmów mitochondrialnych w starzeniu się mózgu i neurodegeneracji, z uwzględnieniem dowodów eksperymentalnych, konkretnych chorób (AD, PD, ALS, HD), biomarkerów oraz możliwości terapeutycznych.
Mechanizmy mitochondrialne
- Dysfunkcja łańcucha oddechowego (ETC) – uszkodzenia kompleksów I–IV prowadzą do zmniejszonej produkcji ATP i wzrostu generacji ROS. Niedobór energii osłabia funkcje neuronów, a nadmiar ROS uszkadza lipidy, białka i DNA (zarówno jądrowe, jak i mitochondrialne). Upośledzenie ETC może być pierwotnym defektem genetycznym (mutacje genów kodujących podjednostki enzymów) lub wtórnym efektem zmian patologicznych (np. agregatów białka α-synukleiny w PD).
- Stres oksydacyjny (ROS/RNS) – mitochodria są głównym źródłem ROS podczas fosforylacji oksydacyjnej. W warunkach dysfunkcji mitochondrialnej równowaga pro- i antyoksydacyjna zostaje zachwiana, co prowadzi do przewlekłego stresu oksydacyjnego. ROS/RNS indukują uszkodzenia białek, lipidów i mtDNA, nasilając dysfunkcje neuronów oraz procesy zapalne. W mechanizmach starzenia i neurodegeneracji nadmierna produkcja ROS jest kluczowym czynnikiem uszkadzającym komórki nerwowe (model oksydacyjnego uszkodzenia).
- Depolaryzacja błony mitochondrialnej (ΔΨm) – utrata potencjału elektrycznego na wewnętrznej błonie mitochondrium wskutek przewlekłego uszkodzenia prowadzi do otwarcia mitochondrialnego kanału błonowego (mPTP) i uwolnienia cytochromu c, inicjując szlak apoptozy komórki. W badaniach opisano, że w modelach ALS czy HD zaburzony jest proces utrzymania ΔΨm, co sprzyja śmierci neuronów (np. w komórkach SOD1G93A stwierdzono obniżony ΔΨm i uwalnianie cytochromu c).
- Kontrola jakości mitochondriów (MQC) – obejmuje biogenezę nowych mitochondriów, dynamikę sieci mitochondrialnej oraz selektywną eliminację uszkodzonych organelli (mitofagię). Centralnym regulatorem biogenezy jest czynnik PGC-1α, który aktywuje geny jądrowe (NRF1/2, TFAM) zwiększając replikację mtDNA i powstawanie nowych mitochondriów. Dynamika mitochondrialna to równowaga fuzji (białka MFN1/2, OPA1) i podziału (Drp1, Fis1, MFF), niezbędna dla wymiany materiału genetycznego i segregacji uszkodzonych fragmentów. Zarówno w starzeniu, jak i w chorobach ND opisano nieprawidłową dynamikę: nadmierny podział prowadzi do pofragmentowanej sieci, a zaburzenia fuzji ograniczają naprawę uszkodzeń.
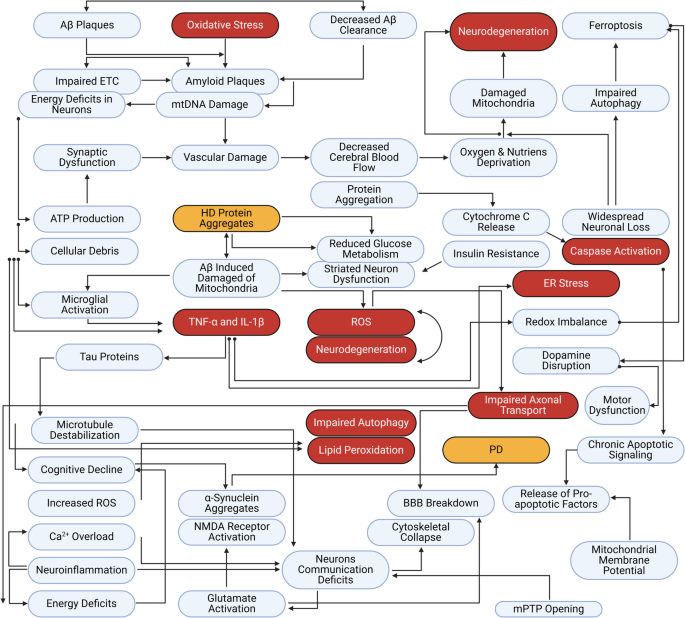
- Mitofagia – proces selektywnego usuwania zużytych mitochondriów. Utrata ΔΨm powoduje akumulację kinazy PINK1 na OMM i rekrutację Parkin, oznaczając uszkodzone mitochondria do autofagosomu (z udziałem p62, Beclin1, LC3). W neurodegeneracji mitofagia jest często zaburzona (np. mutacje PINK1/Parkin w PD obniżają efektywność mitofagii, co powoduje akumulację dysfunkcjonalnych mitochondriów). Poniższy schemat ilustruje wybrane interakcje przyczynowo-skutkowe łączące dysfunkcję mitochondriów z apoptozą i neurodegeneracją:
Podsumowanie mechanizmów: W starzeniu i neurodegeneracji zaburzona jest efektywność OXPHOS (spadek ATP), nasilony jest stres oksydacyjny oraz nieprawidłowa kontrola jakości mitochondriów. W efekcie dochodzi do chronicznej apoptozy neuronów i łagodnej zapadalności („steżenia neurozapalne”) sprzyjających powstawaniu patologii (Bartman i in., 2024; Wadan i in., 2025).
Dowody eksperymentalne
- Badania in vitro: W hodowlach komórkowych z mutantami genów związanych z ND obserwuje się analogiczne defekty mitochondrialne jak u pacjentów. Na przykład komórki neuronalne lub fibroblasty z mutacją PINK1 czy parkiną wykazują gromadzenie uszkodzonych mitochondriów i zwiększony ROS. Modele komórek SH-SY5Y ze zmutowanym SOD1 pokazują obniżone ΔΨm i wzrost aktywności UCP2, co wskazuje na uszkodzenie kompleksu I. Eksperymenty z traktowaniem mitoagresorami (MPTP, rotenon) w linii komórkowej dopaminergicznych neuronów potwierdziły toksyczność wynikającą z hamowania kompleksu I (Lucchesi i in., 2025).
- Modele zwierzęce: Myszom mutacyjnym (np. SOD1^G93A dla ALS, R6/2 i YAC-HD dla Huntingtona) towarzyszą wyraźne anomalie mitochondrialne. W mózgach i rdzeniu tych zwierząt wykazano obniżenie aktywności kompleksów I–IV i zmniejszone stężenie ATP. W PD modelowanej toksycznie (np. MPTP) dochodzi do selektywnej degeneracji neuronów dopaminergicznych przy potwierdzonym niedoborze aktywności kompleksu I w istocie czarnej. Modele genetyczne Huntingtona (myszy R6/2, YAC) ujawniły spadek funkcji kompleksów II–IV, wzrost oksydacyjnego stresu i fragmentację mitochondriów.
- Badania kliniczne: Analizy tkanki mózgowej i płynów ustrojowych od pacjentów ND dostarczają potwierdzeń na poziomie klinicznym. W SN pacjentów z PD stwierdzono ~60% niedobór aktywności kompleksu I w porównaniu z prawidłowymi neuronami. U chorych na ALS fragmenty łańcucha oddechowego (kompleksy I, IV) mają obniżoną aktywność w rdzeniu kręgowym i mięśniach. W AD obniżona jest liczba kopii mtDNA w neuronach i płynie CSF, a aktywność enzymów OXPHOS jest zredukowana nawet przed pojawieniem się zmian amyloidowych. Coraz więcej uwagi poświęca się biomarkerom mitochondrialnym – np. obniżony poziom wolnego mtDNA w płynie mózgowo-rdzeniowym pacjentów AD traktuje się jako wczesny wskaźnik patologii. Pomimo licznych badań, większość dowodów pozostaje niepołączona w jedną spójną przyczynową teorię; wciąż brakuje dużych badań klinicznych oceniających terapie pro-mitochondrialne.
Choroby neurodegeneracyjne
- Alzheimer (AD): W AD obserwuje się dysfunkcję kompleksu IV łańcucha oddechowego i zaburzenia metabolizmu energetycznego neuronów. Komórki z chorobą Alzheimera wykazują podwyższony stres oksydacyjny, zmniejszoną produktywność ATP i zaburzenia homeostazy Ca²⁺, które często pojawiają się przed objawami choroby. Zachwiana jest również dynamika mitochondrialna (zmieniony stosunek fuzji do podziału) oraz mitofagia. Związek z patologią AD pośredniczy „kaskada mitochondrialna” (β-amyloid i tau nasilają dysfunkcję mitochondriów). W próbach zrównoważenia tych defektów stosowano antyoksydanty (np. witaminy, CoQ10), modulatory PGC-1α czy agonisty GLP-1 (liraglutyd), jednak wyniki kliniczne są niejednoznaczne.
- Parkinson (PD): Charakterystyczna dla PD jest utrata neuronów dopaminergicznych i obecność ciał Lewy’ego (agregaty α-synukleiny). Zarówno formy rodzinne (mutacje Parkin, PINK1, DJ-1) jak i sporadyczne (toksyczne agenty MPTP, pestycydy) celują w mitochondria. Zarzuca się im zahamowanie kompleksu I i podwyższenie ROS. Mutacje PINK1/Parkin upośledzają mitofagię, a mutacje PINK1 dodatkowo obniżają potencjał ΔΨm i aktywność kompleksów I/IV. W wynikach badań tkankowych osób z PD wykazano znaczny niedobór aktywności kompleksu I (ponad 60%) w neuronach SN. W sumie dane podkreślają mitochondria jako „węzeł centralny” podatności neuronów PD.
- ALS: W ALS obserwuje się wielopłaszczyznowe zaburzenia mitochondrialne. Mutacje SOD1, TDP-43 czy C9orf72 prowadzą do upośledzenia fosforylacji oksydacyjnej (często kompleks I), zaburzeń przewodzenia elektrycznego ΔΨm i nieefektywnego buforowania Ca²⁺. W modelach zwierzęcych SOD1^G93A stwierdzono obniżenie aktywności kompleksów I, II, IV w rdzeniu kręgowym (już przed wystąpieniem objawów), wiązane ze wzrostem ROS i oksydacją kardiolipiny. Dane z tkanek od pacjentów również potwierdzają spadek ETC w mięśniach i fibroblastach oraz dysocjację kompleksów mitochondrialnych. Cechą ALS jest też dysfunkcja transportu mitochondrialnego w aksonach i zaburzenia dynamiki organelli (m.in. agregacja mitochondriów wokół jądra).
- Huntington (HD): W HD mutacja genu IT15 (mHtt) hamuje funkcje mitochondriów. Zgłoszono redukcję aktywności kompleksów II–IV i spadek produkcji ATP. Mitochondria chorych mają zmniejszoną zdolność buforowania Ca²⁺, są bardziej podatne na depolaryzację i fragmentację. W modelach HD (myszy R6/2, YAC) występuje wyraźny wzrost stresu oksydacyjnego, pofragmentowane mitochondria i obniżona biogeneza mtDNA. HD wiąże się też ze wzmożonym zużyciem ciała (wzrost laktatu), co wskazuje na deficyty energetyczne w mózgu. Obecne dane sugerują, że zaburzenia mitochondriów leżą w centrum patogenezy HD.
Tabela porównawcza mechanizmów i dowodów w chorobach neurodegeneracyjnych:
| Mechanizm / dowody | AD | PD | ALS | HD |
|---|---|---|---|---|
| Geny/mutacje | APP, PSEN1/2, APOE4 | PARKIN, PINK1, SNCA, LRRK2 | SOD1, TDP-43, FUS, C9orf72 | HTT (mHtt) |
| Uszkodzenia ETC | ↓ Kompleks IV, ↓ ATP, ↑ ROS | ↓ Kompleks I, II, IV, ↑ ROS | ↓ Kompleks I (szk.), ↓ II, IV; ↓ ATP | ↓ Kompleks II–IV, ↑ ROS |
| Potencjał błony (ΔΨm) | ↓ ΔΨm | ↓ ΔΨm (PINK1 mut.) | ↓ ΔΨm (SOD1, TDP-43) | ↓ ΔΨm (zaburz. Ca²⁺, linie hodowlane) |
| ROS i oksydacja | ↑ ROS, uszk. DNA/białek | ↑ ROS (neurotoksy, mutacje) | ↑ ROS (SOD1, TDP-43) | ↑ ROS, deficyty enzymów antyoksydacyjnych |
| Dynamika (fuzja/podział) | Zaburzona fuzja/podział | Zaburzona fuzja/podział (Parkin/PINK1) | Fragmentacja mitochondriów | Zaburzona fuzja/podział, fragmentacja |
| Mitofagia | Obniżona lub nieadekwatna | Zaburzona mitofagia (mutacje Parkin/PINK1) | Nadmierna lub nieefektywna mitofagia (SOD1) | Nieokreślona (proponowana niedostateczność) |
| Dowody z badań | ↓ mtDNA w mózgu/CSF, markery (SOD2, cf-mtDNA) | Modele MPTP/rotenon, biopsje SN z ↓CI | Tkanki pacjentów: ↓ kompleks I/IV w rdzeniu, model SOD1^G93A (↓CI/IV) | Biopsje: ↓ II–IV, ↑ laktat; modele R6/2: fragmentacja i ↓ mtDNA |
(Legenda: ↑ – wzrost, ↓ – spadek; wartości cytowane z literatury).
Interwencje terapeutyczne
W ostatnich latach zidentyfikowano wiele strategii ukierunkowanych na mitochondria w ND:
- Leki farmakologiczne: Specjalne antyoksydanty mitochondriotropowe (np. MitoQ, SkQ1) mogą neutralizować ROS bezpośrednio w macierzy mitochondrialnej, co wykazano eksperymentalnie jako neuroprotekcyjne. Agoniści PGC-1α (np. tiazolidinediony, resweratrol) stymulują biogenezę mitochondriów, zwiększając ich liczbę i sprawność. Leki blokujące mPTP (minocyklina, rasagilina) czy modulatory β-amyloidu (metformina, liraglutyd) wykazują w modelach łagodzenie dysfunkcji mitochondriów.
- Interwencje genetyczne: Terapie antysensowne, edycja genowa (CRISPR/Cas9) oraz wektory wirusowe mogą naprawiać mutacje mitochondrialnego DNA (np. delecje mtDNA) lub geny wpływające na funkcje mitochondriów. Wstępne badania z zastosowaniem CRISPR pokazują możliwość naprawy wadliwych genów PINK1 czy POLG, choć w praktyce klinicznej technologia ta wciąż jest we wczesnej fazie rozwoju.
- Modyfikacje metaboliczne: Dieta ketogeniczna i suplementy (córy al. acetoacetate, β-hydroksymaślan, dikarboksylowy kwas octowy – DCA, acetylo-L-karnityna) poprawiają metabolizm mitochondrialny i bioenergetykę. W modelach ALS DCA poprawiał przetrwanie komórek glejowych i funkcje mitochondrialne.
- Zmiana stylu życia: Regularna aktywność fizyczna pobudza PGC-1α przez ścieżkę AMPK, zwiększając biogenezę mitochondriów. Przykładowo mitochondria uzyskane od myszy po intensywnym treningu przy transplantacji do modelu PD poprawiły funkcje OXPHOS i zredukowały patologię choroby. Podobnie uważa się, że ograniczenie kaloryczne stabilizuje funkcje mitochondriów.
- Terapie eksperymentalne: Przeszczep mitochondriów (transfer zdrowych mitochondriów do komórek pacjenta) oraz terapie komórkowe z komórek macierzystych (wydzielające czynniki wzrostu poprawiające mitochondria) są w fazie badań przedklinicznych. Udana transplantacja mitochondriów z komórek młodych do starszych neuronów poprawiła metabolizm energetyczny i przeżywalność komórek w modelach zwierzęcych.
Podsumowanie interwencji: Badane terapie dążą do eliminacji przyczyny (modyfikacja genów, naprawa mtDNA), korekty metabolizmu (dieta, leki pobudzające OXPHOS), oraz wzmacniania obrony antyoksydacyjnej (mito-antyoksydanty). Większość koncepcji wykazała poprawę stanu mitochondriów in vitro lub u zwierząt, jednak brakuje szerokich badań klinicznych potwierdzających skuteczność u ludzi. Często stosuje się też podejście kombinowane, np. jednoczesne podawanie antyoksydantów i suplementów metabolicznych.
Dyskusja
Mimo rosnącej liczby dowodów pozostaje wiele niejasności i wyzwań:
- Przyczynowość vs. skutek: Nie jest jednoznacznie ustalone, czy dysfunkcja mitochondrialna inicjuje procesy neurodegeneracyjne, czy jest ich konsekwencją. Przykładowo w ALS modele SOD1^G93A wykazują silne defekty mitochondrialne, ale u pacjentów z niektórymi mutacjami SOD1 lub TDP-43 obserwuje się zaskakująco zachowane funkcje mitochondriów. Podobnie, w AD niewielkie ilości uszkodzeń mitochondriów występują u osób z prawidłową funkcją kognitywną, co sugeruje wieloczynnikowy charakter choroby.
- Heterogenność kliniczna: Błędy mitochondriów i ich mechanizmy różnią się znacznie między poszczególnymi pacjentami i chorobami. W PD zarówno czynniki genetyczne, jak i środowiskowe wpływają na różne etapy OXPHOS i QC mitochondriów. W związku z tym nie ma uniwersalnego „profile” mitochondrialnego obejmującego wszystkich chorych.
- Przekraczanie bariery krew-mózg (BBB): Większość badań przedklinicznych stosuje bezpośrednie manipulacje w mózgu albo komórki eks vivo. W praktyce farmaceutycznej duże wyzwanie stanowi dostarczenie leków czy terapii genowej przez BBB do neuronów.
- Brak sprawdzonych biomarkerów: Chociaż postulowano różne wskaźniki (cf-mtDNA, SOD2, MMP-2/9), żaden z nich nie jest jeszcze szeroko zwalidowany w dużych kohortach. Konieczne są badania śledzące zmiany biomarkerów mitochondrialnych w czasie choroby i ich korelację z obrazowaniem mózgu.
- Ograniczona skuteczność terapii: Do tej pory żaden duży trial kliniczny leczenia ukierunkowanego na mitochondria (np. podawanie wysokich dawek CoQ10, witaminy E, idebenonu) nie wykazał istotnego zahamowania progresji PD czy AD. Wniosek jest taki, że poprawa parametrów mitochondrialnych na poziomie molekularnym musi być połączona z modyfikacją procesów zapalnych i proteostazy, by przynieść kliniczną korzyść.
Wnioski i luki: Badania literaturowe potwierdzają, że nieprawidłowe funkcjonowanie mitochondriów stanowi ważny czynnik w starzeniu się mózgu i neurodegeneracji (Bartman i in., 2024; Wadan i in., 2025). Mechanizmy takie jak deficyt ATP, stres oksydacyjny, dysbioza dynamiki mitochondrialnej oraz zakłócona mitofagia zostały zaobserwowane we wszystkich analizowanych chorobach. Opracowano liczne strategie terapeutyczne celujące w te patologie mitochondriów, jednak ich sukcesy w badaniach klinicznych są dotąd skromne. Kluczowe potrzeby przyszłych badań to: (i) lepsze modele odzwierciedlające ludzką patogenezę, (ii) identyfikacja wiarygodnych biomarkerów mitochondrialnych u ludzi oraz (iii) kompleksowe terapie łączące ochronę mitochondriów z modulacją neurozapalania. Pomimo istniejących kontrowersji, konkluzja jest jasna – zrozumienie i wsparcie funkcji mitochondrialnej pozostają obiecującą drogą do modyfikacji przebiegu chorób neurodegeneracyjnych.
Bibliografia
- Bartman S, Coppotelli G, Ross J. Mitochondrial dysfunction: a key player in brain aging and diseases. Curr Issues Mol Biol. 2024;46(3):1987–2026.
- Wadan AHS, Shaaban AH, El-Sadek MZ, Mostafa SA, Nady R, Mehanny SS, et al. Mitochondrial-based therapies for neurodegenerative diseases: a review of the current literature. Naunyn Schmiedebergs Arch Pharmacol. 2025;398:11357–11386.
- Wang S, Liao Z, Zhang Q, Han X, Liu C, Wang J. Mitochondrial dysfunction in Alzheimer’s disease: a key frontier for future targeted therapies. Front Immunol. 2025; (data/dokument, epublication).
- Lucchesi M, Biso L, Bonaso M, Longoni B, Buchignani B, Battini R, et al. Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. Int J Mol Sci. 2025;26(9):4451.
- Cunha-Oliveira T, Montezinho L, Simões RF, Carvalho M, Ferreiro E, Silva FSG. Mitochondria: A promising convergent target for the treatment of amyotrophic lateral sclerosis. Cells. 2024;13(3):248.
- Joshi DC, Chavan MB, Gurow K, Gupta M, Dhaliwal JS, Ming LC. The role of mitochondrial dysfunction in Huntington’s disease: Implications for therapeutic targeting. Biomed Pharmacother. 2025;183:117827.
- Di Lorenzo R, Zecca C, Chimienti G, Latronico T, Liuzzi GM, Pesce V, et al. Reliable new biomarkers of mitochondrial oxidative stress and neuroinflammation in cerebrospinal fluid and plasma from Alzheimer’s disease patients: a pilot study. Int J Mol Sci. 2025;26(16):7792.
Opracowanie: Rafał Klasiński / Fundacja Independent Institute of chemical Processes